Tutorial videoes
The video below describes the operation of multilocus phylogeny pipeline in PhyloSuite.
The video below describes the operation of multilocus phylogeny pipeline in PhyloSuite.
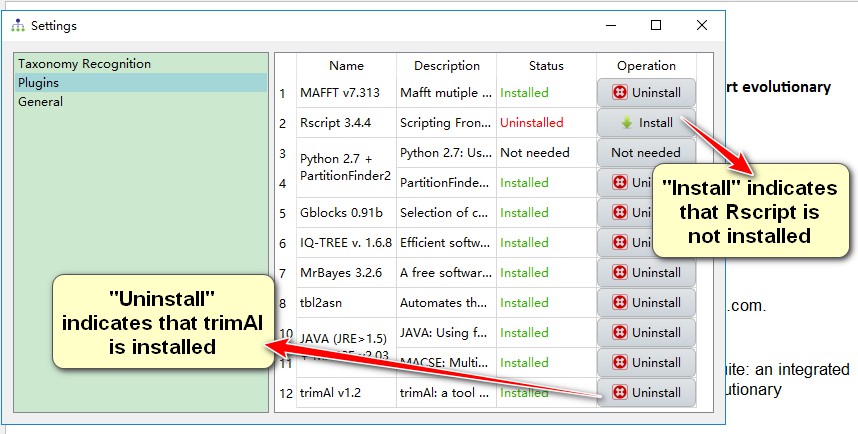
There are two ways to configure the plugins:
When using Windows and Mac OSX systems, you can download PhyloSuite_xxx_Win32/64_with_plugins.rar and PhyloSuite_xxx_Mac_with_plugins.zip from https://github.com/dongzhang0725/PhyloSuite/releases, respectively. All plugins, except Python 2.7, Perl 5, Java, Rscript, HmmCleaner and trimAl (MAC) are included.
When extracting sequences, you should use different settings for different data-types. PhyloSuite has five predefined data-types, Mitogenome, general, cox1, 16S and 18S, but you can easily add your own data-type extraction settings, suited to your data.
Here I will demonstrate the process of making the extraction settings for the mitochondrial genome data-type.
PhyloSuite: from data acquisition to phylogenetic tree annotation (muti-gene dataset)

In order to make it easier for users to get started, here we briefly introduce the use of PhyloSuite using an example of all available Monogenea (class) (Platyhelminthes) mitochondrial genomes (mitogenomes).
Author’s Note: To save space, the contents duplicated with our single-gene tutorial will not be detailed in this article.
PhyloSuite: from data aquisition to phylogenetic tree annotation (single gene tree building)

Please see http://phylosuite.jushengwu.com/dongzhang0725.github.io/documentation/#4-3-3-1-Brief-example.
Chinese homepage link: http://phylosuite.jushengwu.com/
Please see http://phylosuite.jushengwu.com/dongzhang0725.github.io/installation/.
Please see http://phylosuite.jushengwu.com/dongzhang0725.github.io/PhyloSuite-demo/how-to-configure-plugins/.